Skip to main content
Search
Search
Microboids
Sharing Knowledge improves Knowledge... Knowledge should come at as less cost as possible.
Blog
Posts
More…
Posts
Showing posts from March, 2017
Show All
Posted by
Varun C N
March 31, 2017
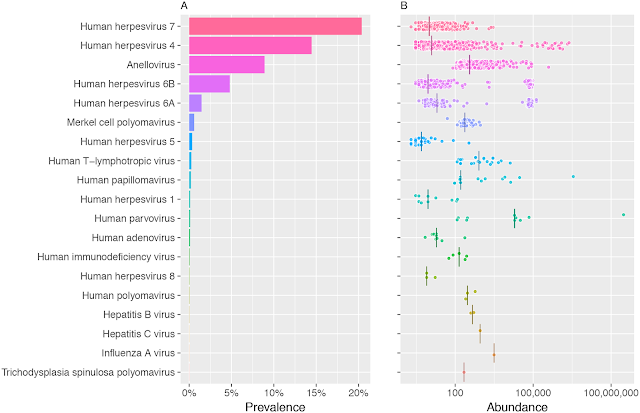
Human Blood Virome
Posted by
Varun C N
March 22, 2017
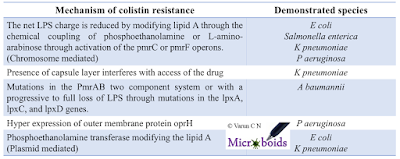
MCR gene variants
Posted by
Varun C N
March 17, 2017
Bedaquline and Delamanid resistance
Posted by
Varun C N
March 07, 2017
WHO global PPL for antibiotic development
Posted by
Varun C N
March 01, 2017
BtB# 12- Why is ID a Requirement in Clinical Microbiology
Newer Posts
Older Posts
Home